|
|
Eine einheitliche Aussage ist nicht
möglich; vielmehr ist von einm weiten Spektrum an
Ursachen bzw. Assoziationen auszugehen  , welche sich in ihren pathoklinischen
Auswirkungen überschneiden können. , welche sich in ihren pathoklinischen
Auswirkungen überschneiden können. |
Zum
besseren Verständnis von Ätiologie und Pathogenese der
FOHA sei erwähnt, daß ein Organ z.B. das Ovar sowohl
primär, als auch sekundär beteiligt sein kann.
Die wesentlichen Ätiologievorstellungen der FOHA
werden nun im folgenden genauer erläutert: |
- Genetische Ursachen
|
Die
Biosynthese der wichtigsten ovariellen Sexulasteroide D
4-Androstendion, Testosteron, Östradiol und Progesteron kann auf vielfältige Weise gestört werden.
Jüngere Untersuchungen (Lobo et al, 1996  , Franks et al, 1997 , Franks et al, 1997 ) machen
deutlich, daß genetisch bedingte intraovariellen
Dysfunktionen der Thekazelle und auch der Granulosazelle eine mögliche Rolle spielen, die auch
erklärlich machen könnten, warum derzeitig angewandte
Therapieprinzipien oftmals nicht die erhoffte kurative
Wirkung zeigen . Die Erhöhung der intraovariellen
Androgenkonzentration kann zudem die Morphologhie des
Ovars wesentlich beeinflussen . ) machen
deutlich, daß genetisch bedingte intraovariellen
Dysfunktionen der Thekazelle und auch der Granulosazelle eine mögliche Rolle spielen, die auch
erklärlich machen könnten, warum derzeitig angewandte
Therapieprinzipien oftmals nicht die erhoffte kurative
Wirkung zeigen . Die Erhöhung der intraovariellen
Androgenkonzentration kann zudem die Morphologhie des
Ovars wesentlich beeinflussen . |
| Die FOHA
kann primär oder sekundär durch die höhere Anzahl an
antralen Follikeln, durch eine höhere Anzahl an
Thekazellen pro Follikel oder durch eine höhere
Freisetzung an C19-Steroiden pro Thekazelle
(Gilling-Smith et al, 1994 ) erklärt
werden. Die erhöhte Präsenz an
klein-antralen Follikeln pro Ovar ist eines der
wesentlichen morphologischen Charakteristika des meist
mit der FOHA assoziierten PFO II-III0
(» "PCOS") .
Pathomorphologisch kann die FOHA in seltenen Fällen auch
mit einer Hyperthekosis assoziiert sein.
Übergänge zwischen PFO III0
und Hyperthekosis sind möglich.
|
|
|
| Neben den
dominanten endokrinen Faktoren (Gonadotropine,
Sexualsteroide) spielen zahlreiche Wachstums- und
Differenzierungsfaktoren vor Ort für das Ovar
(Geisthövel., 1992 ) eine bedeutende Rolle. Diese
nicht-gonadotropen, nicht-steroidalen Faktoren können
die basalen Ovarialfunktionen, wie z.B. das follikuläre
Rekruitment oder die Selektion des dominanten Follikels
etc. wesentlich mit modulieren und bei pathologischen
Prozessen wie bei der FOHA/PFO von Bedeutung sein. Sie
können - zumindest theoretisch - primäre ovarielle
Dysfunktionen hervorrufen, aber auch durch systemische
Störfaktoren sekundär beeinflußt sein. Daß derartige
primäre, intraovarielle, von der Gonadotropinachse
unabhängige Faktoren evident sein müssen, läßt sich
durch die klinische Erfahrung ableiten, daß oft eine
jahrelange Suppression der Gonadotropinsekretion durch
z.B. orale Kontrazeption keine wesentliche Veränderung
der pathomorphologischen Komponente mit sich bringt. Am
intensivsten ist das IGF-System für das menschliche Ovar
(s.Rev. Geisthövel, 1992 ; s.Rev. Giudice,1997 ) untersucht worden: In
Ebene B findet sich eine ausführliche Darstellung des
Themas:
|
Physiologische
IGF-Effekte am menschlichen Ovar
IGF:
Theka-Granulosa-Zell-Kompartment
Insulin-, IGF
I-Rezeptoren
IGF-Bindungsproteine
Zusammenfassung:
Pathologie des IGF-Systems beim "PCOS"
|
| Systemische
Veränderungen des IGF-Systems beeinflussen auch die
Wirkung des Wachstumhormons (human growth hormone, hGH).
So kann bei Suppression der IGF-Bindungsproteine ein
Hyposomatotropismus die Folge sein, der im Zusammenhang mit
metabolischen Veränderungen steht; postmenopasual spricht man auch von Somatopause,
um darauf hinzuweisen, daß der Abfall des hGH mit dem
Altern der Frau auch Mitursache des sog. Menopausalen
Metabolischen Sydnroms sein kann. |
|
|
| Inhibine
und Activine sind Protein-Dimere der
Transforming-Growth-Factor-b Familie. Durch
Kombinationen der verschiedenen Untereinheiten sind zwei
Inhibine A und B und drei Aktivine bekannt. Man nimmt an,
daß ovarielle Inhibine die FSH-Sekretion bremsen und
Activine diese steigern. Ihre die Follikelfunktion
modulierenden Eigenschaften scheinen nicht nur auf
endokrinen, sondern auch auf parakrine Interaktionen zu
beruhen. Insgesamt scheinen die Effekte des
Inhibine-Activine-Systems in der Pathogenese der FOHA von
Bedeutung zu sein: Inhibine-Activine-Systeme |
|
|
| Nach der Yen-Theorie (Yen, 1991 ) kann durch eine Mehrsekretion des adrenalen
DHEAS über eine Dysregulationsspirale von
verschiedenen Zwischenstationen [ Fettgewebe,
Konversion (Östronbildung ),
Hypothalamus-Hypophyse (sek. LH/FSH )] eine
sekundäre FOHA induziert werden. Es sei hier
daraufhingewiesen, daß jede der konsekutiven
Zwischenstationen auch als primäre Einstiegsstelle (z.
B. Adipositas, Dysfunktion der hypothalamo-hypohysären
Achse) gelten kann. |
Adrenale Ätiologie
Extraglanduläre
Aromatisierung
Sexualsteroid-Metabolismus:
Fettgewebe
Pathologische
LH-Pulsatilität
Theka-Granulosa-Zell-Kompartment
|
|
|
| Eine der wesentlichen Dysfunktion im Rahmen der
FOHA ist die chronische, azyklische Anhebung des
LH/FSH-Quotienten (Arroyo et al, 1997 ). Es gibt Hinweise für eine intrinsische
funktionell oder genetisch bedingte Störung des
zentralen Dopaminmetabolismus (s.Rev. Lobo,1996 ), der eine primär
zentrale Ursache der LH/FSH-Erhöhung sein kann.
Von größerer Bedeutung sind wohl sekundäre
LH-Anhebungen durch periphere endokrine Dysfunktionen,
wie z.B. druch eine chronische Anhebung der
zirkulatorischen Östronkonzentration . Wie auch immer verursacht, führt eine
chronisch-azyklische LH-Übersekretion zur dominanten
Stimulation des ovariellen Theka-Zell-Kompartments mit
entsprechender sek. FOHA. Nochmals zusammengfaßt sind
folgende Dysfunktionsbereiche, die in Ebene B erläutert
werden, zu beachten:
|
Hypothalamo-hypophysäre
Funktionsstörung
Extraglanduläre
Aromatisierung
Sexualsteroid-Metabolismus:
Fettgewebe
Pathologische
LH-Pulsatilität
Schnittstelle: Adipositas
- FOHA
|
|
|
| Das SHBG ist der wichtigste zirkulatorische
Bindungsträger des Testosterons, das seine periphere
Funktionen im wesentlichen nur in freier Form ausüben
kann. Eine Senkung des SHBG geht konkordant mit einer
Anhebung des freien, biologisch aktiven Testostoterons
einher. Allein durch eine Senkung des SHBG kommt es zu
einer systemischen Erhöhung der Androgenität. Insofern
spielt das SHBG eine besondere Rolle als
Androgenitätsdeterminante. Dieses Thema wird in Ebene B
ausführlich dargestellt: |
Physiologie des SHBG
Bindungscharakteristika
von C19-/C18
-Steroiden und ihren Bindungsproteinen
Folgen einer
Hypo-SHBG-ämie
Ursachen: Hypo-SHBG-ämie
Ursachen:
Hyper-SHBG-ämie
|
|
|
| Kurz soll auf die Physiologie des Glucose-Insulin-Stoffwechsels
eingegangen werden. Nach der enteralen
Hydrolyse von Disacchariden (durch Disaccharidasen)
erfolgt die Aufnahme von Monosaccariden (Glucose,
Fructose) in die Zirkulation. Bei der pankreatischen
Perfusion wird der postprandiale Monosaccharidanstieg
registriert, und es erfolgt die exophytische Freisetzung
des Insulins aus der b -Zelle des Pankreas, wodurch die
Glukohomöostase (Glykolyse, Glukose-uptake) wieder
hergestellt wird. |
| Unter pathologischen Bedingungen, insbesondere
im Rahmen einer peripheren Insulinresistenz, kann es
(kompensatorisch) zur Hyperinsulinämie kommen. Seit
bekannt geworden ist, daß die Hyperinsulinämie
in der Genese der FOHA eine besondere Rolle spielt
(Barbieri et al, 1988 ), ist die
Auseinandersetzung mit dem Glucose- und
Insulinmetabolismus im Rahmen der FOHA ein wichtiger
Bestandteil wissenschaftlicher und klinischer
Untersuchungen geworden (Geisthövel et al, 1994 ; Nestler et
al, 1991; Nestler und
Jakubowicz, 1996 ). Insulin
wirkt nahezu identisch wie IGF-I als Ko-Gonadotropin
, nur mit deutlich geringerer Effektivität als
dieses am Theka-Granulosa-Zell-Kompartment. So nimmt man
an, daß eine chronische, zirkulatorische Insulinanhebung
zu einer Steigerung der thekalen
Androgenbiosysnthese durch Aktivierung der P450c17a
(Nestler
und Jakubowicz, 1996 ) führt.
Desweiteren beeinflußt Insulin in erheblichem Maße die
hepatische Bindungsprotein-Synthese (z.B.
IGFBP-1, SHBG, GHBP) , wodurch die systemische Androgenität eine
deutliche Steigerung erfährt (Nestler et al, 1991 ). |
| Zudem scheint die Hyperinsulinämie auch an der
Genese des Hyposomatotropismus durch die Suppression der hepatischen
IGF-BP 1 Sekretion und der Hyper-, Dyslipidämie
beteiligt zu sein. Die Effekte des Insulins am
Ovar werden durch ortsständige Insulinrezeptoren, aber
auch durch die homologen IGF-I-Rezeptoren vermittelt (s. Rev. Geisthövel et al, 1992 ). Die Insulinresistenz
beim PCOS scheint selektiv nur das Fettgewebe und den
Skelettmuskel bzw. den Glukosetransport und die Glykolyse
zu betreffen (Peiris et al, 1989 ); diese
selektiven Unterschiede könnten auch erklären, warum
die Hyperinsulinämie trotz der zugrundeliegenden
Insulinresistenz dennoch Störungen auf ovarieller
(Androgensekretion ) und hepatischer (SHBG-Sekretion¯ ,
IGF-BP-Sekretion¯ ) Ebene induzieren kann. Die Ursachen der
Hyperinsulinämie - Insulinresistenz sind vielfältig; für den Gynäkologen sind in
diesem Zusammenhang
|
| physiologische Formen im Rahmen der: |
- Pubertät
- Schwangerschaft und im
- fortgeschrittenen Alter
|
| von pathologischen zu unterscheiden bei: |
- Adipositas
- Non-insulin dependent diabetes
mellitus (NIDDM=Typ II Diabetes) und bei der
- FOHA.
|
| Zur weiteren Vertiefung des Verständnisses zur
Ätiologie der Hyperinsulinämie wird daraufhingewiesen,
daß die Hyperinsulinämie primär im Rahmen einer
genetisch bedingten pankreatischen Übersekretion oder kompensatorisch-sekundär im Rahmen
einer peripheren Insulinresistenz hervorgerufen sein kann. |
|
|
| Schon seit der Erstbeschreibung durch Stein und
Leventhal (Stein und Leventhal, 1935 ) ist die Assoziation
von Adipositas, kutaner, androgenisierender Symptomatik und
"polyzystischen Ovarien" bekannt. Dies ist
gerade in jüngster Zeit von zunehmender Bedeutung, da
die Adipositas weltweit zahlenmäßig ansteigt, so daß
sogar von einer epidemischen Ausbreitung
gesprochen wird (Bouchard, 1991 ). Als Ursachenkomplex
wird die weltweite Verwestlichung (besonders:
Amerikanisierung) des Lebensstils angesehen mit einer
ständigen Zunahme des Fettanteils in der Nahrung und einer immer häufiger zu beobachtenden körperlichen
Immobilität , die bereits in der Kindheit festzustellen ist; auch durch den Einfluss des
familliären Umfelds zusammen mit genetischen Komponenten
ergibt sich, dass bei Kindern eine positive Korelation
der Adipositas-Prävalenz und des Body Mass Index (BMI)
mit dem BMI der Eltern besteht;
weiterhin scheint auch der Grad der schulischen
Ausbildung eine Rolle zu spielen. Man nimmt an (s.
Bouchard, 1991 ), daß ca 36% der Frauen in Nordamerika
Übergewicht und Adipositas haben und daß 25 bis 50% der
erwachsenen amerikanischen Bevölkerung versuchen
abzunehmen oder diätetische Maßnahmen ergriffen haben.
Die chronische Adipositas muß in die Reihe der Eßstörungen
eingeordnet werden. Multifaktoriell verursacht stellt sie
sich in zahlreichen heterogenen Phänotypen mit einem
weiten Spektrum klinischer Symptome dar, das von gesunden,
fertilen und höchstens kosmetisch beeinträchtigten
Frauen bis zu einem schwerwiegenden,
endokrin-metabolischen, mitunter das gesamte Leben
bestimmenden Adipositassyndrom reicht. |
| Definierte mit Adipositas assoziierte
Erkrankungen, die durch die Mutation eines
einzelnen Gens verursacht sind, stellen die Ausnahme dar
und fallen mehr in den Aufgabenbereich des Pädiaters
und Internisten. |
| Man geht eher davon aus, daß in der weitaus
überwiegenden Mehrzahl der Krankheitsbilder oligo-
oder polygenetische Störungen mit anderen Faktoren, z.B. Dysfunktionen
der Nahrungs- und Ernergieregulation(z.B. im Rahmen einer Hyperleptinämie)
interagieren; diese verschiedenen Faktoren können dann
innerhalb eines gestörter Regelkreises mit somatischer Inaktivität, gestörtem
Eßverhalten und Depressionen dann zur
phänotypischen Ausprägung der Adipositas führen. |
| Im Fettgewebe besteht ein großes
Aromatasepotential , welches die Umwandlung von Androgenen und
schwach androgen wirksamen Androgenpräkursoren in
Östrogene (Östron, E1) ermöglicht. Hieraus
resultiert eine chronische Stimulation der Gonadotropin
releasing hormone (GnRH) -abhängigen hypophysären
LH-Sekretion, welche wiederum eine vermehrte Stimulation
des ovariellen Theka-Kompartimentes mit verstärkter Androgenproduktion bewirkt.
Dies ist eine der Schnittstellen zwischen Adipositas
und FOHA . |
| Außerdem ist die Adipositas mit einer Abnahme
der Insulinsensitivität und konsekutiv mit
einer Hyperinsulinämie (s.o.) sowie mit einem Hyposomatotropismus
und
einer Hypo-SHBG-ämie assoziiert, weitere Schnittstellen
zwischen Adipositas und FOHA . |
| Von besonderer Bedeutung ist, daß die
Adipositas über die schon genannten Störformen hinaus
in ein Netzwerk schwerwiegender Komorbiditäsfaktoren
eingebunden sein kann, wozu auch das sog. Metabolische
Syndrom zählt,
das ein hohes Risiko für eine Mikro-, Makroangiopathie
und und letztlich für die Koronare Herzkrankheit darstellt.
Dies ist von besonderer Bedeutung, wenn die Adipositas
seit Kindheit besteht und einen lebenslangen Prozeß
darstellt . Durch die schon erwähnten Schnittstellen
zwischen Adipositas und FOHA kann auch diese mit einem
Risiko für die KHK assoziiert sein  Differentialdiagnostisch müssen noch die
schweren, genetisch bedingten, häufig mit Fehlbildung
einhergehenden Adipositassyndrome abgegrenzt werden. Differentialdiagnostisch müssen noch die
schweren, genetisch bedingten, häufig mit Fehlbildung
einhergehenden Adipositassyndrome abgegrenzt werden.
|
|
|
| Wie schon mehrfach erwähnt wird für die
phänotypische Expression der FOHA und assoziierter
Störungen bereits eine Reihe genetisch bedingter
Dysfunktionen verantwortlich gemacht. Oligo-, polygenetische
Ätiologie in Kombination mit anderen nicht-genetisch
bedingten Faktoren (z.B. Ernährung, Umwelt) sind
anzunehmen. Es sind schon eine ganze Reihe an Studien
durchgeführt worden; so fanden sich verschiedene Gendefekte
bzw. Polymorphismen bei FOHA ; bei den Untersuchungen
fanden sich aber auch: Gene ohne Mutationsnachweis bei
FOHA . |
| Von großer Bedeutung ist es zu verstehen,
daß unterschiedliche Ursachen phänotypisch ähnliche
klinische Ausdrucksbilder induzieren: so findet sich
eine FOHA auf dem Boden einer polyfollikulären
Ovarstruktur sowohl
bei intraovariellen, genetisch verursachten Enzymstörungen
als auch
bei systemischen Dysfunktionen, z.B. durch
Hyperinsulinämie infolge einer z.B. fehlernährungsbedingten
Adipositas. Umso mehr können Kombinationen
solcher Dysfunktionen zum Erscheinungsbild der FOHA
führen. Oftmals sind Ursache und Wirkung nicht
auseinanderzuhalten. Weiterhin ist zu verstehen, daß die
o.g. komplexen intraovariellen
Dysregulationsmeachnismen unabhängig vom
Gonadotropinstimulus wirksam und daher therapeutisch
schwer beeinflußbar sind . |
|
|
| An dieser Stelle sollte noch die
Aktivitöätserhöhung der 5a -Reduktasen
Erwähnung finden, die ätiologisch eigentlich nicht der
FOHA zuzuordnen ist (s.aber unten!), jedoch vom
klinischen Standpunkt aus gesehen nicht von der FOHA
zutrennen ist; daher sei sie hier kurz erwähnt. |
| Die
zu den androgen-abhängigen Störungen zählende "Idiopathische"
androgenisierende Symptomatik kann durch eine
Aktivitätsanhebung der intraepidermalen 5a -
Reduktasen Typ I und Typ II bedingt
sein, spezifische Enzyme, die das zirkulierende
Testosteron in das androgenetisch aktivste C19-Steroid,
das Dihydrostestosteron umwandeln,
das sich an den spezifischen Androgen-Rezeptor des
Erfolgsorgans bindet. Der Typ I scheint mehr in den
Talgdrüsen (Akne) und der Typ II scheint mehr in den
Haarfollikeln (Hirsutismus, Alopezie) lokalisiert zu sein
(Rittmaster, 1995 ). Bei dieser rein intraepidermal lokalisierten
Störung fehlen systemische Dysfunktionen. Der Begriff
"idiopathisch" ergibt sich, da unter klinischen
Bedinungen eine Diagnostik der intrakutanen 5a
-Redukatse-Aktivität nicht durchgeführt werden kann. |
| Selbst schwerer Hirsutismus kann ein rein
lokales, letzendlich kosmetisches Symptom sein! Es muß aber daraufhingewiesen
werden, daß möglicherweise auch im Ovar 5a
- Reduktasen wirksam sind , wodurch sich evt. auch Therapieindikationen
für die FOHA ergeben .
|
| |
|
|
In dem folgenden Schema wird der
Stellenwert der FOHA/PFO II°-III° innerhalb des
Hyperandrogenismus der Frau dargestellt:Vorschlag zur Systematisierung der
Begriffsbestimmungen
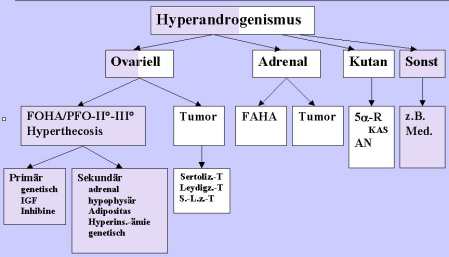
Legende:* FAHA: funktionelle adrenale Hyperandrogenämie;
5a -R: 5a -Redukatse Aktivitäts-Erhöhung; KAS: kutane
androgenisierende Symptomatik; AN: Acantosis nigricans;
Med.: anabol-androgenisierende Medikamente; Sertoliz.-T:
Sertolizell-Tumor; S.-L.z.-T:
Sertoli-Leydigzell-Mischtumor.
|
